Raquel viajó a Bélgica en busca de un tratamiento para su hijo: «Escribí a hospitales de varios países para que nos aceptasen en un ensayo clínico»

ENFERMEDADES

Nanook, de 16 años, fue uno de los cerca de cien niños reclutados en todo el mundo para el estudio EXIST-1, que tenía como protagonista un fármaco que ahora mismo ya se administra en España
20 oct 2025 . Actualizado a las 05:00 h.Él no se acuerda de todo, pero Nanook pasó una gran parte de su infancia en Bélgica. Hasta allí se trasladó con su familia en numerosas ocasiones desde el 2010, cuando solo tenía unos meses de vida. Hasta el año pasado, porque si bien el estudio concluyó oficialmente en el 2014, él siguió recibiendo el medicamento. A día de hoy, gracias a él y a otros casi cien niños de todo el mundo que fueron reclutados, este se encuentra aprobado y se comercializa en la Unión Europea y, por lo tanto, también en nuestro país. Se trata del everolimus, un fármaco inmunosupresor que se administra contra los astrocitomas de células gigantes (tumores benignos de crecimiento lento) que padecen algunos pacientes con esclerosis tuberosa, un tastorno genético poco frecuente por una mutación en los genes TSC1 o TSC2. Se estima que hay entre 4.000 y 2.300 personas diagnosticadas en nuestro país.
«Cuando nos dieron el diagnóstico, todo lo que me estaban diciendo en el hospital me parecía terrorífico. Estuvimos ingresados durante días y me puse a buscar en internet información sobre lo que le estaba pasando a mi hijo», recuerda Raquel Galavís, madre de Nanook. El astrocitoma, un tumor cerebral que le estaba afectando en ese momento, implicaba un riesgo inminente. Así fue como encontró un estudio clínico que estaba a punto de cerrarse, en el que reclutaban niños con un diagnóstico reciente «y que además tuviesen el astrocitoma». Y sin pensarlo dos veces, «escribí a todos los hospitales de diferentes países que lo estaban realizando (en aquel momento, aún no se encontraba ninguno de España)».
Todos le dijeron que no, que debía residir en ese país, menos la neuróloga del Hospital UZ de Bruselas, Anna Jansen, «que me dijo que sí». El estudio se llamaba EXIST-1, era un estudio en fase III y proporcionó la base científica para que se aprobase la medicación. «Es el primer tratamiento a día de hoy para los astrocitomas de células gigantes asociados a la esclerosis tuberosa, sobre todo cuando los pacientes no son operables», matiza Raquel.
Se calcula que hasta un 90 % de los niños que padecen esta enfermedad presentan manifestaciones a nivel del sistema nervioso central, como la epilepsia, las alteraciones cognitivas, problemas de comportamiento y autismo, tal como indica la Asociación Española de Esclerosis Tuberosa. De esta forma, entre las anomalías neurológicas que pueden acomapañar a estas manifestaciones, se encuentran: los túberes corticales, nódulos subependimarios y astrocitomas subependimados de células gigantes (conocidos bajo las siglas SEGA) y hamatorimas cerebrales.
Los SEGA son una preocupante manifestación neurológica de la esclerosis tuberosa por el potencial riesgo que suponen a la hora de originar cuadros agudos de hidrocefalia y aumento de la presión intracraneal, por obstrucción del drenaje del líquido cefalorraquídeo.
El ensayo clínico
Al principio, la familia viajaba cada trece días a Bélgica. A los tres meses, cada veinte. «Se iban esparciendo cada vez más las visitas, pero siempre estábamos monitorizados con resonancias magnéticas que asegurasen que ese astrocitoma no crecía». Después de un año se desveló qué grupo de pacientes estaba tomando placebo (era un ensayo de doble ciego) y Nanook era uno de ellos. «Su astrocitoma no creció, pero tampoco fue a menos».
Ahí, le proponen continuar en una fase abierta, recibiendo la medicación en uso compasivo. Es decir, el fármaco sigue en estudio y no autorizado para una indicación específica, pero se puede administrar a los pacientes que en los que se observa beneficios. «Duró varios años y, tan pronto como empezó a tomar el fármaco, el astrocitoma se redujo de 2,5 a 1,4 centímetros, que es como está ahora. Existen efectos adversos, pero no tan graves como para que, en una balanza de riesgo y beneficio, no gane este último».
Un 22 % de los ensayos clínicos aprobados en nuestro país en el 2024 tenían como protagonistas las enfermedades raras.
La última vez que estuvieron en Bélgica fue el año pasado. «Aunque lo que era el estudio para la aprobación del medicamento había terminado, siguieron monitorizando a algunos pacientes que quisimos para ver el efecto a largo plazo de la medicación», explica. Y a pesar de todos los días que estuvo ingresado en el hospital, tanto Raquel como su marido removieron cielo y tierra para hacer planes que lo evadiesen. «Creo que no tiene una experiencia traumática, al contrario, conserva recuerdos bonitos de la ciudad».
Voluntarios del presente para pacientes del futuro: la radiografía de los ensayos clínicos en nuestro país
España es el país de Europa donde más investigaciones de este tipo se han aprobado en el 2024, siendo el cáncer protagonista del 39 % de los estudios realizados. En general, la representación de las mujeres suele ser inferior a la de los hombres: un tercio de todos los participantes, una problemática sobre la que ponen el foco diferentes instituciones
España se volvió a convertir en el 2024 en el país con la mayor capacidad de atracción de investigación clínica de medicamentos de la Unión Europea. Según el Registro Español de Estudios Clínicos (REec), la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) autorizó el año pasado un total de 930 estudios, alzándose como la agencia reguladora europea que más ensayos clínicos ha aprobado. Detrás de estas cifras existe un trabajo coordinado y continuo entre diferentes agentes que hacen posible que esa terapia o fármaco se acabe aprobando y administrado en aquellos pacientes que lo necesiten.
Un ensayo clínico es una investigación en seres humanos para demostrar la eficacia o seguridad de un medicamento. Así lo define, de forma resumida, Juan Estévez, jefe de la División de Ensayos Clínicos de la AEMPS. Para que puedan ponerse en marcha, deben cumplirse una serie de criterios. El primero es la autorización por parte de la autoridad competente. «En este caso, somos nosotros los que la otorgamos. Hay un proceso de evaluación científica por parte de la agencia y de ética, por parte de un comité», explica. Pueden llevarse a cabo por iniciativa comerical o no. «Detrás de esta última puede haber un investigador, una fundación u otro tipo de promotor; son el 20 % de los ensayos, el resto, son propuestos por la industria farmacéutica».
Las distintas fases de los ensayos clínicos
La investigación clínica pasa por distintas etapas. En la primera, conocida como fase I, participan unos pocos voluntarios (de veinte a treinta, aproximadamente), para confirmar que la administración del fármaco es segura. En la fase II comienzan los análisis de seguridad de esas dosis y, por lo tanto, el grupo de participantes también aumenta, pudiendo llegar a unas 200 o 300 personas. Se estudia también la seguridad con las dosis exploradas. Cuando el propósito es la confirmación de la eficacia de un fármaco, el ensayo se considera de fase III. En este caso ya participan miles de personas y el objetivo ya es obtener datos precisos para la solicitud de una autorización de comercialización.

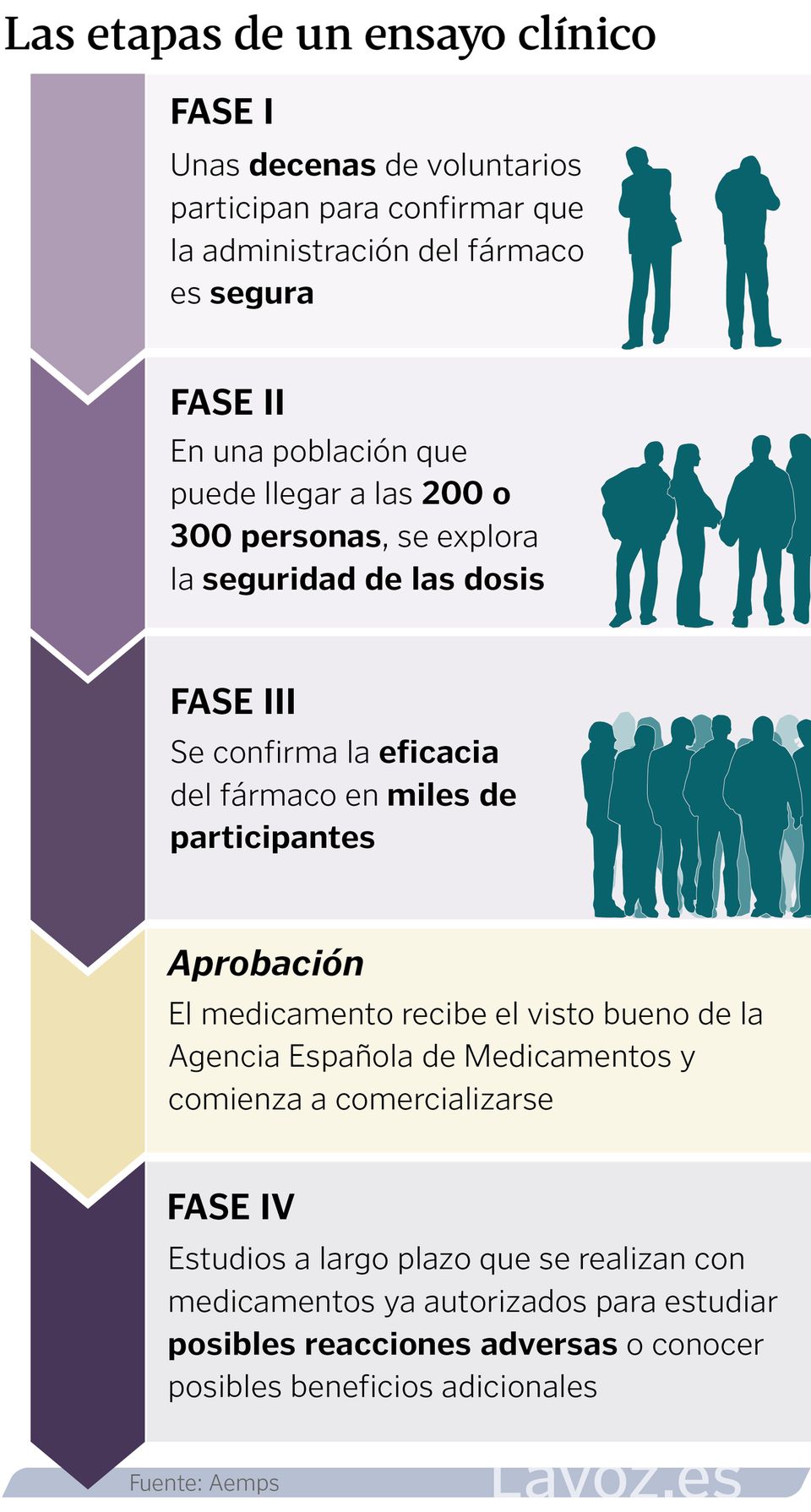
Puede decirse que los ensayos más particulares son los de fase IV. Estos ya son a largo plazo con medicamentos que están autorizados, para estudiar posibles reacciones adversas o incluso conocer posibles beneficios adicionales. «Al final, en farmacología, cuanto más amplías la población, que es lo que sucede cuando se autoriza, cuentas con más posibilidades para poder buscar problemas de seguridad que no hayan aparecido hasta el momento, o que busques eficiencias en el sistema, mejorando, por ejemplo, dosis o pauta», amplía Estévez.
Con todo, el jefe de la División de Ensayos Clínicos de la AEMPS matiza que «al final lo determinante no es el título que se le dé al estudio, si lo llamas de fase II o III, porque algunos incluso pueden estar en la frontera entre una y otra, sino que esté bien controlada la seguridad de los sujetos y que los datos que se generen sean lo suficientemente robustos».
Qué sucede si el ensayo clínico sale bien (o mal)
«El proceso de generación de datos y de autorización es complejo», avanza Estévez. «Para intentar resumirlo, cabe decir que, con cada ensayo clínico que finaliza, independientemente de cómo se haga, se tiene que generar un informe de resultados porque esto nos permite, si funciona bien, entenderlo mejor y si ha salido mal, también, para después tenerlo en cuenta en futuras autorizaciones de ensayo clínico».
Para presentar una autorización de comercialización, hay varias vías. «La más conocida es la centralizada: se presenta a la Agencia Europea de Medicamentos (EMA), la cual coordina el procedimiento y las agencias nacionales aportan evaluadores que estudian estos dosieres. Si bien existen otros procedimientos que se utilizan cada vez menos, como el nacional, que englobaría a un medicamento que solo se quiere autorizar en nuestro país; o el reconocimiento mutuo, autorizar el fármaco solo en determinados países de la Unión Europea. Con todo, son procesos que, con la legislación actual, se están quedando muy reducidos, porque lo que se busca es que toda la población europea se pueda beneficiar de nuevos fármacos».
Por lo tanto, poniendo el foco en la vía centralizada, una vez que la EMA da el visto bueno porque el balance entre el beneficio y el riesgo de ese fármaco es positivo, se otorga la autorización. «Después, comienza el proceso de financiación y precio en cada país. En el caso de España, es competencia del Ministerio de Sanidad y en función del tipo de fármaco y su presupuesto, se puede tardar más o menos en dar acceso a ese fármaco. Pero este proceso, ya no es competencia de la AEMPS», detalla.
Si se da cualquier tipo de complicación en el ensayo clínico, la agencia tiene capacidad para paralizarlo y, en el peor de los casos, también de suspenderlo. «Pero lo que suele ocurrir es que es el propio promotor el que paraliza el tratamiento o reclutamiento. En función de la gravedad de lo ocurrido, incluso podría romperse el ciego para que se sepa qué se ha dado a los pacientes o qué no», cuenta el experto. En un ensayo de doble ciego, ni los participantes ni los investigadores saben qué tratamiento o intervención están recibiendo o administrando hasta que el estudio finaliza. Así, un grupo recibe la terapia que se está evaluando, mientras que el otro, un placebo que se siente idéntico a la terapia original. Se realiza de esta forma para minimizar el sesgo, evitando que las expectativas influyan en los resultados. Las conclusiones son más objetivas y fiables.

Líderes europeos en investigación para tratar el cáncer
España lidera la investigación de medicamentos contra el cáncer en Europa. De los 930 ensayos clínicos a los que dio luz verde la AEMPS en el 2024, 350 estaban destinados a tratar el cáncer, suponiendo un 39 % de los estudios autorizados. Charo García Campelo, jefa de servicio de Oncología del Complexo Hospitalario Universitario de A Coruña (Chuac) y responsable del grupo de investigación de Oncología del Instituto de Investigación Biomédica de A Coruña (Inibic), los define como «un acto de enorme generosidad por parte de esa persona hacia futuros pacientes, porque si bien este accede a nueva posibilidad terapéutica, también a la incertidumbre de no saber cómo funcionará lo que le estamos proponiendo». Lo explica desde la unidad de ensayos clínicos situada en el hospital coruñés. «Ahora mismo, aquí, tenemos en torno a 190 abiertos, en distintos tumores sólidos: cáncer de mama, colorrectal, de pulmón, melanoma... Pero también en diferentes etapas, desde fase I a fase III, así como estudios observacionales».

Conforme se sube en fase, aumentan los participantes de cada estudio, pero se reduce el número de ensayos. «Los más numerosos que tenemos son fase III: en el 2024, fueron 86; en fase II, 64; y en fase I, tenemos 8. Estos últimos son los más complejos porque son los que implican más labores en términos de seguridad y de obtención de la dosis eficaz y tolerable para un paciente, son los que se hacen en esta unidad específica. El resto, se hacen en consulta convencional». Además, la doctora comenta que en la unidad de fase I los tumores más frecuentes son aquellos en los que han fracasado la mayoría de alternativas terapéuticas disponibles.
La participación en un ensayo clínico es muy rigurosa. «Es lógico que esté sometido a tanto control. Existen unos criterios de inclusión que deben ser cumplidos de forma global por ese paciente en concreto». Asimismo, Campelo remarca que el proceso de información tiene que ser claro. «El paciente tiene que estar informado de lo que supone en términos de posible eficacia, toxicidad y beneficios».
Una participación de mujeres menor
Las mujeres participan menos en ensayos clínicos. Hace unas semanas, un estudio llevado a cabo por el Centro Nacional de Investigaciones Cardiovasculares (CNIC) en colaboración con la Sociedad Española de Cardiología, revisó el uso de betabloqueantes tras haber sufrido un infarto de miocardio. De él partía un subestudio, con el que se espera seguir adelante, sobre los efectos de esta medicación en mujeres. Óscar Prada, cardiólogo del Complexo Hospitalario Universitario de A Coruña (Chuac), en declaraciones a La Voz de la Salud, remarcaba que «había que poner el foco con especial interés» en la participación de las mujeres en este tipo de estudios, «para tener una información mucho más robusta» sobre la administración de algunos fármacos: «Con algunos tratamientos puede ocurrir que la dosis necesaria sea diferente según el sexo, y por lo tanto, también se mejoraría el efecto de ciertos medicamentos. Al final, no somos iguales biológicamente».

El jefe de la División de Ensayos Clínicos de la AEMPS confirma que «es un tema que preocupa y que tenemos que abordar». Señala que ya existen diferentes iniciativas por parte de la agencia e incluso del Ministerio de Sanidad, para tratar «la diversidad» en los ensayos clínicos. «Aquí se incluye, efectivamente, el tipo de participación que ha existido de mujeres y hombres, y si se han tenido en cuenta los posibles efectos que pueden aparecer según la dosis».
«Hay una participación reciente que asegura que la menor participación de mujeres en ensayos clínicos es un problema, ya que la inclusión de población femenina en ensayos clínicos se sitúa en torno a un 30 %. Esto es un problema, porque la mayoría de la investigación, históricamente, ya sea preclínica y clínica, se ha hecho en hombres. Y de ahí, asumimos muchas conclusiones en torno a dosis, eficacia de fármacos, tolerancia de una determinada estrategia terapéutica o incluso la calidad de vida que esta puede proporcionar», aborda la oncóloga Campelo. Si bien a día de hoy, la población masculina y femenina difiere en muchos de los aspectos comentados. «En cómo se metabolizan los fármacos, cuál es la dosis más adecuada o la forma de percibir la toxicidad; por todo esto, la participación de mujeres tiene que ser un tema prioritario, tanto en oncología como en otras especialidades».
En cuanto a las razones que pueden llegar a explicar esa infrarrepresentación de la población femenina en los ensayos se engloban causas de exclusión históricas de las mujeres en los estudios y la falsa creencia de que los cuerpos masculinos conformaban un modelo representativo para ambos sexos. Si bien la doctora Campelo añade otras que se suman a la lista: «Hay distintos factores que pueden explicar el bajo reclutamiento de las mujeres en ensayos clínicos. Esto está publicado, registrado y analizado. Uno de ellos es que, a veces, la propia mujer rechaza la participación en un ensayo clínico porque este exige mucha visita al hospital, muchos procedimientos que consumen tiempo, y sus obligaciones laborales y familiares, de conciliación, pueden llevarla a rechazar la opción». En su opinión, los profesionales sanitarios como ella deben animar a que participen y «ayudarlas a que tengan las herramientas necesarias para que su vida personal no se vea tan impactada como para decir que no a la investigación».